





El pasado mes de noviembre, la FDA publicó un “Informe de cumplimiento y calidad de dispositivos médicos”. A continuación hago un resumen de los puntos que más me llamaron la atención:
1- La FDA ha aumentado la cantidad de inspecciones de manufacturadores de dispositivos médicos, especialmente en el extranjero
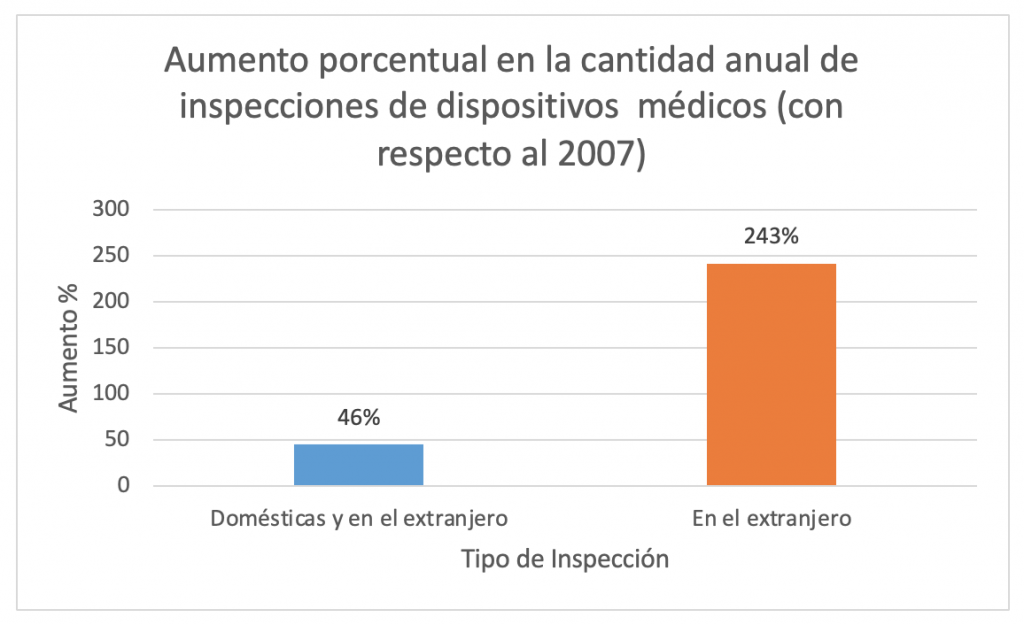
Desde el 2007 a la fecha, la FDA ha aumentado en un 46% la cantidad anual de inspecciones de manufacturadores de dispositivos médicos (tanto dentro como fuera de los Estados Unidos), en tanto las inspecciones sólo en el extranjero han aumentado un 243%.Es decir, las inspecciones en el extranjero han aumentado 5.3 veces más que las inspecciones totales.

2-La FDA ha aumentado su enfoque durante las inspecciones en el cumplimiento de las Partes 803 y 806 del CFR
21
CFR Parte 803: MDR- Reportes de dispositivos médicos
Los inspectores de la FDA se han enfocado en verificar que las empresas hayan reportado los eventos adversos (MDRs) de sus dispositivos médicos de acuerdo con la Parte 803 del CFR (Código de Regulaciones Federales por sus siglas en inglés).
MDR – Medical Device Reporting, es el mecanismo de la FDA para recibir reportes de eventos adversos significativos, de forma que puedan ser detectados y corregidos de forma rápida. La FDA requiere que las compañías de dispositivos médicos reporten dos tipos de incidentes: 1) los que ocasionan o podrían ocasionar la muerte del paciente), y 2) los incidentes que pueden tener efectos serios sobre la salud del paciente.
Este mayor enfoque en las inspecciones en la Parte 603 ha contribuido a que el reporte de MDRs a la FDA se haya duplicado desde el 2009.
21
CFR Parte 806: Informes de correcciones y retiros
LaParte 806 del CFR requiere que los manufacturadores informen a la FDA de cualquier corrección o retirada (recall) voluntaria del mercado de un dispositivo médico.
Del mismo modo, la FDA se ha estado enfocando durante sus inspecciones en el cumplimiento de este apartado de la regulación, lo cual ha resultado en un aumento del 50% en el número de recalls voluntarios desde el 2009.Esto quiere decir que las compañías han decidido con más frecuencia hacer recalls voluntarios de productos potencialmente defectuosos.


3- La FDA ha adoptado un enfoque
basado en el riesgo para identificar productos específicos que presentan problemas
potenciales en el mercado
La FDA monitorea el mal funcionamiento de los dispositivos médicos y las tendencias de cumplimiento de las compañías, y cuando identifica problemas con la calidad del dispositivo o el fabricante, inspecciona estas compañías para reducir la posibilidad de problemas similares y recurrentes.Entre los dispositivos que la FDA ha identificado con este tipo de problemas se encuentran las bombas de infusión, los desfibriladores externos automatizados (AED por sus siglas en inglés)y los dispositivos de terapia de radiación.


Entre las acciones tomadas para mejorar el cumplimiento de las regulaciones y la calidad de estos productos, la FDA:
- Incrementó la cantidad de inspecciones a las compañías manufacturadoras, emitiendo Cartas de Advertencia (Warning Letters) en los casos que encontró incumplimientos
- Eliminó el uso del 510 (k) como mecanismo de aprobación previa a la entrada al mercado para los desfibriladores externos
En un blog anterior explicamos la vía de aprobación del 510 (k): 510(k)vs PMA ¿Cuál es la diferencia?
Como resultado de estas y otras acciones específicas para cada tipo de dispositivo, la FDA logró una reducción significativa en la cantidad de recalls y MDRs con respecto a años anteriores:
| Tipo de dispositivo | Reducción en cantidad de recalls | Reducción en cantidad de MDRs |
| Bombas de infusión | 56% | 82% |
| Desfibriladores externos | 70% | 27% |
|
Dispositivos de terapia de radiación |
80% | ————– |
4. La FDA ha adoptado un en foque más interactivo con las empresas con problemas de cumplimiento regulatorio
Este enfoque interactivo consiste, particularmente, en que el personal de la FDA revisa las respuestas al formulario 483 (que se usa para notificar a las empresas de condiciones objetables al final de una inspección) y proporciona ala compañía comentarios sobre las acciones correctivas propuestas.
Cuando sea posible, la FDA considera que este puede ser un enfoque más eficaz para lograr planes de acción correctiva más oportunos y efectivos, así como monitorear el progreso hacia la remediación.
Este enfoque más interactivo ha dado como resultado una disminución en el número anual de Warning Letters, pero sin detrimento de la corrección de las violaciones a las regulaciones que se han encontrado durante las inspecciones.
Por su parte, las Warning Letters se han concentrado en las empresas que tienen violaciones graves o que no logran implementar o dar seguimiento a su plan de acción correctiva de manera adecuada.
*****
Le dejo por aquí el link al reporte completo: MedicalDevice Enforcement and Quality Report-November 2018





