





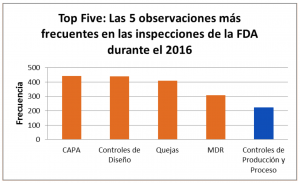
En los primeros meses de este año, hemos estado analizando el ‘Top Five’ de las principales observaciones de la FDA durante las inspecciones a los manufacturadores de dispositivos médicos: “Las 5 principales observaciones de la FDA para los manufacturadores de dispositivos médicos en el 2016”. Finalizamos esta serie de blogs con los 483 al apartado 820.70 Controles de Producción y Proceso, el cual fue el quinto apartado con más observaciones en el 2016:

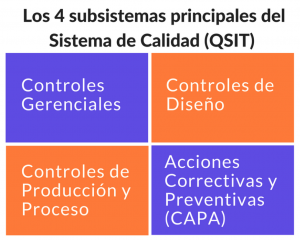
De acuerdo con el QSIT (Quality System Inspection Technique, la técnica de inspección de la FDA), Controles de Producción y Proceso es uno de los 4 subsistemas principales del sistema de calidad, por lo tanto siempre es inspeccionado durante las visitas de la FDA:

¿Y cómo escoge el inspector de la FDA el o los procesos a inspeccionar? El QSIT establece los criterios que el inspector debe seguir para seleccionar un proceso:
- Indicadores de problemas en el proceso provenientes del subsistema de CAPA
- El uso del proceso para la manufactura de dispositivos con nivel de riesgo más alto (por ejemplo dispositivos Clase III)
- El grado de riesgo del proceso para causar fallos en el dispositivo
- La falta de familiaridad y experiencia de la compañía con el proceso (por ejemplo procesos nuevos)
- El uso de proceso en la manufactura de múltiples dispositivos
- La variedad de tecnologías usadas en el proceso (por ejemplo procesos complejos)
- Procesos que no hayan sido cubiertos durante las inspecciones previas


En este blog revisaremos las 5 principales observaciones de la FDA en el 2016 para el apartado 820.70 Controles de Producción y Proceso, las cuales constituyen el 81% de las observaciones de acuerdo con el siguiente diagrama de Pareto:
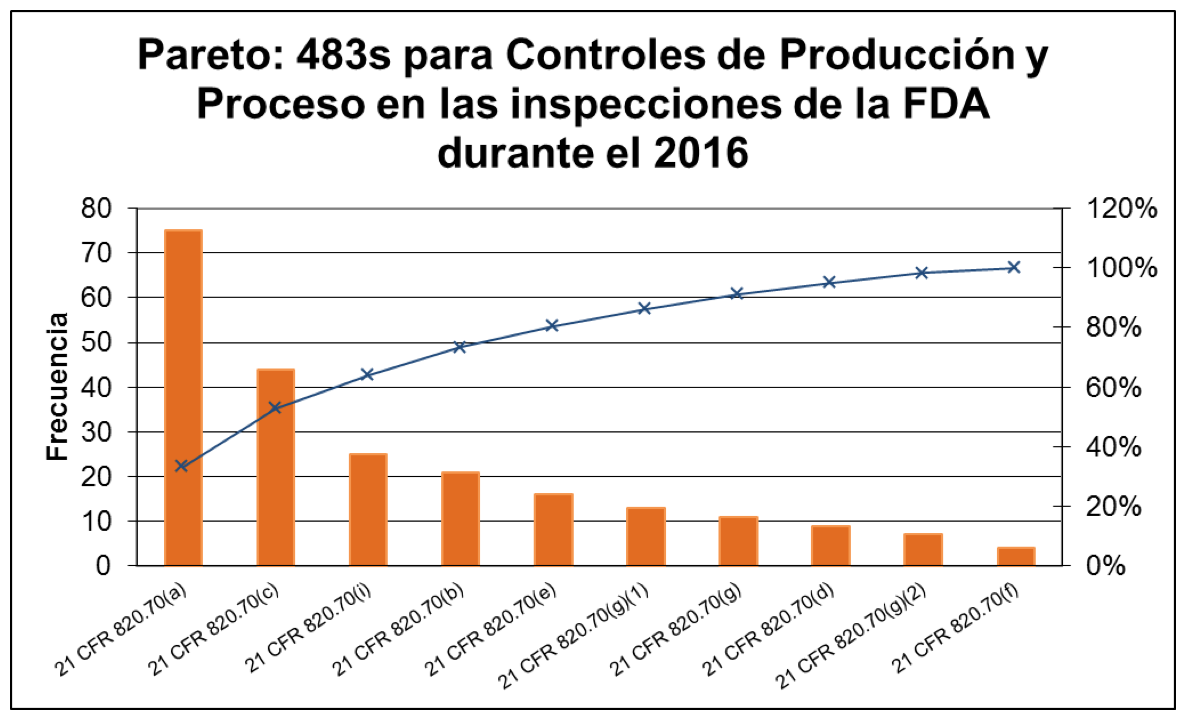

- 21 CFR 820.70(a) – Deficiencias en los procesos de producción, o en los procedimientos escritos para el control de proceso (75 observaciones, 34%) – Las observaciones contra este apartado se debieron a que:
- Los procesos de producción no fueron desarrollados, realizados, controlados o monitoreados para garantizar que el dispositivo cumpla con las especificaciones; o bien
- Los procedimientos escritos que describen los controles de proceso necesarios para asegurar la conformidad con las especificaciones no han sido establecidos de forma adecuada.
- 21 CFR 820.70(c) – Deficiencias en los procedimientos de control ambiental (44 observaciones, 20%) – Los procedimientos para controlar las condiciones ambientales no han sido establecidos de forma adecuada.
- 21 CFR 820.70(i) – Deficiencias en la validación del software usado para procesos automatizados y en la documentación de la validación del software (25 observaciones, 11%) – Las observaciones contra este apartado fueron porque:
- Las actividades y resultados de la validación de software, sistemas para computadoras o procesamiento automatizado de datos, usados como parte del proceso de producción o del Sistema de Calidad, no han sido documentadas de manera adecuada.
- El software usado como parte del proceso de producción o del Sistema de Calidad no ha sido validado adecuadamente para su uso previsto de acuerdo con un protocolo establecido.
- 21 CFR 820.70 (b) – Deficiencias en los procedimientos de cambios en producción y proceso (21 observaciones, 9%) – Los procedimientos para realizar cambios a las especificaciones, métodos, procesos o procedimientos no han sido establecidos de forma adecuada.
- 21 CFR 820.70 (e) – Deficiencias en los procedimientos de control de la contaminación (16 observaciones, 7%) – Los procedimientos para evitar la contaminación de los equipos o el producto con sustancias que puedan tener un efecto adverso en la calidad del producto no han sido establecidos de forma adecuada.
*******************************
Este es el último blog del “Top Five”, los 5 apartados de la regulación con más observaciones de la FDA para los manufacturadores de dispositivos médicos durante el año 2016. Si se perdió alguno de los otros blogs le dejo por aquí los links: CAPA, Controles de Diseño, Quejas, y MDR.





