


El año pasado publicamos por primera vez el resumen de las principales observaciones de la FDA durante las inspecciones realizadas a los manufacturadores de dispositivos médicos durante el año fiscal (FY) 2015. El año fiscal de la FDA es el periodo comprendido entre el 1 de octubre y 30 de setiembre del siguiente año.
La FDA acaba de publicar su resumen anual para el FY 2016, por lo cual les presentamos de nuevo las principales observaciones durante este periodo. Esta información es importante, pues nos permite identificar las posibles áreas de enfoque de la FDA durante sus inspecciones, y verificar si estamos en total cumplimiento con los requerimientos de la agencia.
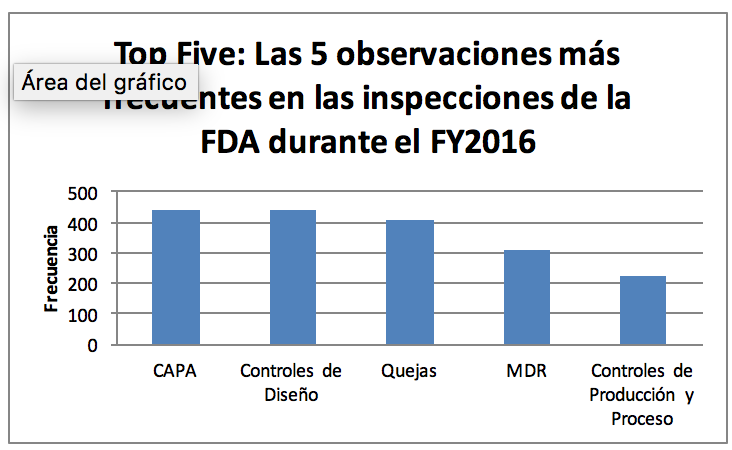 El siguiente es un resumen de los principales requerimientos de la FDA en relación con cada uno de estos cinco elementos de la regulación.
El siguiente es un resumen de los principales requerimientos de la FDA en relación con cada uno de estos cinco elementos de la regulación.
 21 CFR 820.100 CAPA- 443 Observaciones.
21 CFR 820.100 CAPA- 443 Observaciones.
En el FY2016, CAPA fue el proceso con mayor cantidad de observaciones. El sistema de Acciones Correctivas y Preventivas, cuando está bien implementado y es efectivo, es el mecanismo para detectar problemas en el sistema de calidad y llevarlo de nuevo a conformidad.
Este sistema es uno de los más importantes desde un punto de vista regulatorio, por lo cual siempre es inspeccionado por la FDA. Si durante la inspección se identifican no conformidades que no hayan sido detectadas o solucionadas por el sistema de CAPA, la FDA cuestionará la adecuada implementación y efectividad de este sistema por parte del manufacturador.
 CFR 820.30 Controles de Diseño- 441 Observaciones.
CFR 820.30 Controles de Diseño- 441 Observaciones.
La FDA da especial importancia a la fase de diseño, pues la calidad, seguridad y efectividad de un dispositivo se validan durante esta fase. La FDA ha indicado que un alto porcentaje de retiradas de mercado o recalls se deben a problemas de diseño.
La regulación requiere controles de diseño para los dispositivos clase III y II, y para algunos dispositivos clase I. Algunos de los principales requerimientos de la regulación son: establecer procedimientos escritos para el control de diseño, realizar la revisión, verificación y validación del diseño, y documentar todo el proceso de diseño del dispositivo en el DHF (Design History File).
 21 CFR 820.198 Quejas- 409 Observaciones.
21 CFR 820.198 Quejas- 409 Observaciones.
El manejo de las quejas recibidas por el manufacturador es uno de los sistemas más importantes para la FDA. Las quejas pueden provenir de varias fuentes: los centros hospitalarios, los médicos, o directamente de los pacientes.
Las quejas proporcionan información de las fallas del producto en el mercado, por lo cual deben ser investigadas rápidamente. Las regulaciones requieren que todas las quejas, incluso las recibidas oralmente, sean investigadas y respondidas de forma expedita. Adicionalmente, el manufacturador debe mantener registros de las quejas recibidas por parte de los usuarios del dispositivo médico.
Finalmente, las quejas tienen que evaluarse para determinar si son eventos reportables a la FDA de acuerdo con la parte 21 CFR 803 Medical Device Reporting.
 21 CFR 803 MDR (Medical Device Reporting)- 309 Observaciones
21 CFR 803 MDR (Medical Device Reporting)- 309 Observaciones
El manufacturador está obligado a reportar a la FDA las muertes o lesiones serias que el dispositivo haya causado o pueda haber causado durante su uso. Estos eventos adversos pueden estar relacionados con fallas en el producto, mal funcionamiento, un diseño inadecuado, defectos de manufactura, problemas de etiquetado o incluso errores del usuario.
El sistema de MDR permite a la FDA monitorear los eventos adversos relacionados con los dispositivos una vez que están en el mercado, y ordenar acciones de campo si lo considera necesario (un “recall” o retirada de mercado del producto).
 21 CFR 820.70 Controles de producción y Proceso ˗222 Observaciones
21 CFR 820.70 Controles de producción y Proceso ˗222 Observaciones
Este es un apartado central de la regulación pues define los requerimientos de funcionamiento para el “corazón” de una planta de manufactura: el proceso productivo en sí. Describe los requerimientos de la agencia para que los manufacturadores de dispositivos médicos controlen la producción y el proceso de manufactura.
Entre ellos se tiene, por ejemplo: establecer procedimientos operativos estándar (SOP’s), que controlen los métodos de producción, contar con personal entrenado, monitorear y controlar los parámetros de proceso, y establecer mecanismos de control ambiental cuando sea requerido.
*******************************
Si usted no está muy familiarizado con el detalle de los requerimientos de cada uno de estos elementos de la regulación, los próximos blogs le pueden ser útiles pues estaremos analizando con más detalle las observaciones de la FDA para cada uno por aparte.