




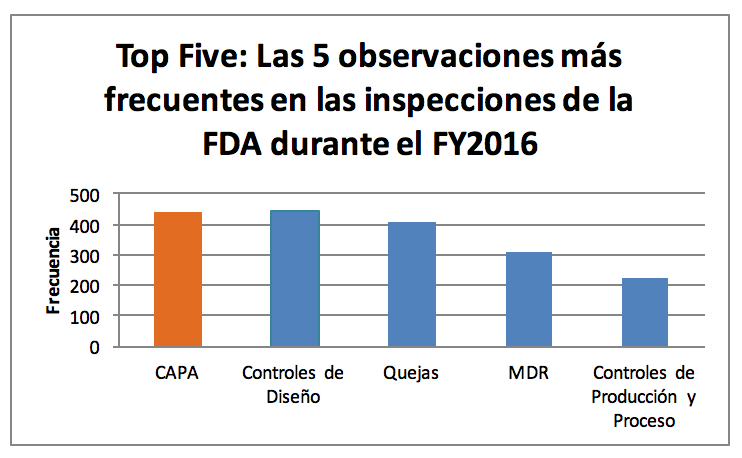
Como vimos en un blog anterior: “Las 5 principales observaciones de la FDA para dispositivos médicos durante el año fiscal 2016”, con un total de 344 observaciones, CAPA fue el proceso que más recibió 483s por parte de la FDA en el 2016:

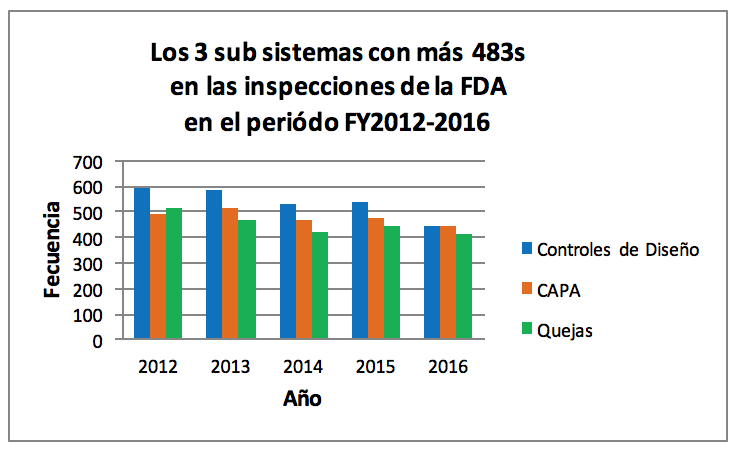
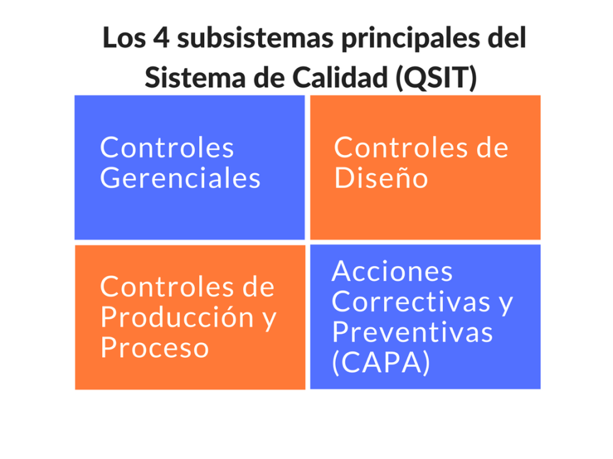
● El subsistema de CAPA es uno de los más inspeccionados por la FDA. De acuerdo con el QSIT (la técnica de inspección de la FDA), CAPA es uno de los 4 subsistemas principales del sistema de calidad, siendo estos: 1) Controles gerenciales, 2) Controles de diseño, 3) Controles de producción y proceso y 4) Acciones Correctivas y Preventivas (CAPA).


● El sistema de CAPA es el mecanismo de “auto reparación” del sistema de calidad. La misión de CAPA es detectar problemas o deficiencias en el sistema de calidad y llevarlo de nuevo a conformidad. Es por esto que, si durante la inspección, la FDA encuentra no conformidades que no hayan sido detectadas o solucionadas por el sistema de CAPA, cuestionará la adecuada implementación y efectividad de este sistema por parte del manufacturador.

Entre las principales deficiencias que puede presentar un sistema de CAPA, y a los cuales hay que prestar especial atención antes de una inspección de FDA tenemos los siguientes:
1. Problemas en el Sistema de Calidad no detectados y resueltos por CAPA. Como mencionamos anteriormente, si durante la inspección se encuentran problemas en el Sistema de Calidad que no han sido ni siquiera detectados por el subsistema de CAPA, serpa interpretado por la agencia como una implementación inadecuada.
2. Falta de prontitud en la implementación de las acciones correctivas y preventivas.
Si bien no hay un tiempo definido por la regulación para la implementación de las acciones correctivas y preventivas, la expectativa de la FDA es que el manufacturador las implemente con la mayor prontitud (“timeliness”) posible.
3. Falta de utilización de herramientas de Análisis de Causa Raíz.
El proceso de CAPA es sistemático, no debemos saltar a las acciones correctivas de los problemas basados en la intuición o la experiencia previa. Para lograr una solución efectiva de los problemas del Sistema de Calidad, la investigación debe aplicar herramientas ingenieriles de “Root Cause Analysis” (RCA), como un mapeo de procesos, un diagrama de Ishikawa, un DMAIC u otro tipo de herramientas dependiendo del grado de complejidad del problema.
4. Recurrencia de problemas en el Sistema de Calidad.
La recurrencia, o aparición de manera de repetitiva de los mismos problemas en el Sistema de Calidad, es un indicativo para la agencia de que el proceso de CAPA no está bien implementado y no es efectivo en resolver los problemas de calidad del manufacturador.

