





Si bien la mayoría de profesionales en industria médica están familiarizados con el requerimiento regulatorio de validar los procesos de producción antes de iniciar a manufacturar producto “vendible”, muy pocos conocen la guía internacional para validación de procesos en la cual se basan los procedimientos de prácticamente todas las compañías de dispositivos médicos.
La “validación de proceso” es un término usado en la industria para indicar que un proceso ha sido sometido a un escrutinio tal, que prácticamente se puede garantizar que el resultado del mismo (es decir, el producto o dispositivo médico) cumplirá con los requerimientos de calidad establecidos.
La “Process Validation Guidance GHTF/SG3/N99-10” fue publicada en el 2004 por GHTF (“Global Harmonization Task Force”), el cual fue un equipo especial de trabajo para la armonización global de las regulaciones para la industria de dispositivos médicos. En la actualidad, el grupo se llama IMDRF (“International Medical Device Regulators Forum”) y agrupa a las agencias regulatorias de países con mercados muy importantes, como los Estados Unidos, la Unión Europea, China, Rusia, Australia, Brasil, Japón y Canadá.


La “Process Validation Guidance” es la que define las conocidas tres fases que utilizamos en industria médica para la validación de un proceso:
1) IQ (Calificación de la Instalación o “Installation Qualification”) – Consiste en una calificación inicial del equipo usado y la provisión de los suministros que este requiera (por ejemplo, aire comprimido, suministro de agua, suministro eléctrico, entre otros).
2) OQ (Calificación Operacional o “Operational Qualification”) – Es una demostración de que el proceso producirá resultados aceptables y el establecimiento de los límites (“worst case” o “peor caso”) de los parámetros de proceso.
3) PQ (Calificación del Desempeño o “Performance Qualification”) – Consiste en el establecimiento de la estabilidad de proceso a largo plazo.
Durante el PQ habitualmente se toman muestras de tres lotes de producción. La razón para usar tres lotes no tiene ningún fundamento estadístico, sin embargo es el estándar aceptado por la FDA.
Otra contribución importante de esta guía es el árbol de decisión para la validación de proceso que se muestra en la siguiente figura:
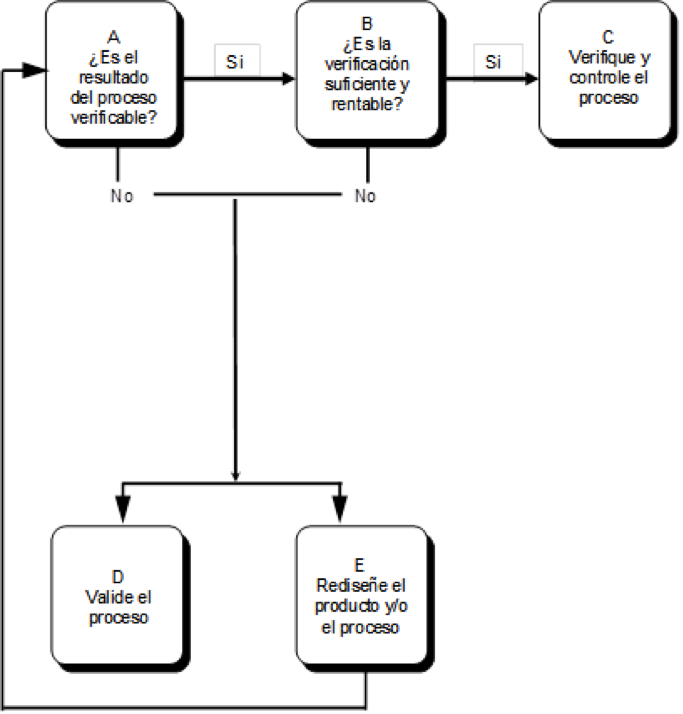

El manufacturador debe considerar si el resultado del proceso puede ser verificado al 100% mediante un monitoreo o medición subsecuente del dispositivo (A). Esto es realizable en el caso de inspecciones 

Adicionalmente, debe evaluarse si la verificación al 100% es suficiente por sí misma para eliminar los riesgos inaceptables en el dispositivo y si es una solución efectiva en términos de costos, es decir, rentable para el manufacturador (B). Si es así, el resultado debe ser verificado y el proceso debe ser apropiadamente controlado (C).
Si el resultado del proceso no es 100% verificable, entonces la decisión apropiada es validar el proceso (D). Por ejemplo, los siguientes procesos deben ser validados, debido a que no es posible realizar una verificación al 100%:
- Procesos de esterilización
- Condiciones ambientales de cuarto limpio
- Procesos de sellado de empaque estéril
- Procesos de tratamiento térmico
- Procesos de moldeo de plástico por inyección
El Anexo A de la guía establece las herramientas y métodos estadísticos que se puedan usar para la validación de procesos. Se mencionan por ejemplo el muy utilizado diseño de experimentos (DOE, “Design of Experiments”, y los estudios de capacidad de proceso.

Finalmente, y regresando al árbol de decisión para la validación de proceso, el manufacturador puede considerar la alternativa de rediseñar el producto o proceso para reducir la variabilidad (E) hasta tal punto que sea posible realizar una simple verificación al 100% (C) .

