







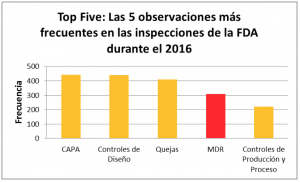
En enero de este año iniciamos una serie de blogs relacionados con “Las 5 principales observaciones de la FDA para los manufacturadores de dispositivos médicos en el 2016”. El presente blog está dedicado al reporte de eventos adversos a la FDA o Medical Device Reporting (MDR). Con 294 observaciones, MDR fue el cuarto apartado de la regulación con más 483s:

El sistema de MDR es una herramienta de vigilancia posterior a la comercialización (postmarket surveillance), que la FDA utiliza para monitorear el desempeño de los dispositivos médicos en el mercado. Cada año, la FDA recibe varios cientos de miles de reportes de muertes, lesiones severas y mal funcionamiento, los cuales se sospecha están relacionados con dispositivos médicos. El sistema de MDR permite a la FDA detectar problemas de seguridad potencialmente relacionados con los dispositivos médicos en tiempo real.


El reporte de eventos adversos a la FDA generalmente no se realiza desde las plantas de manufactura, sino desde las oficinas corporativas. En un blog anterior ¿Qué es Medical Device Reporting?, revisamos los principales requerimientos de los tipos de reporte y el tiempo máximo para reportar a la FDA.
Las principales observaciones de la FDA en el 2016 fueron contra el apartado 803.17– ¿Cuáles son los requerimientos para desarrollar, mantener e implementar procedimientos escritos de MDR? Con 294 observaciones, las deficiencias en el procedimiento escrito de MDR fueron las observaciones más frecuentes:
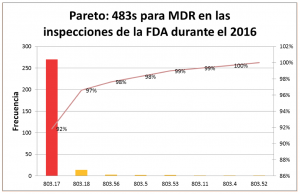

A continuación revisamos las principales observaciones contra el apartado 803.17:
- 21 CFR 803.17 – Deficiencias en el procedimiento escrito de MDR (146 observaciones) No se han desarrollado, mantenido o implementado procedimientos escritos de MDR.
- 21 CFR 803.17(a) (1) – Falta de un sistema para evaluación de eventos (27 observaciones) El procedimiento escrito de MDR no incluye un sistema interno que permita la identificación, comunicación y evaluación oportuna de los eventos que pueden estar sujetos a reporte a la FDA.
- 21 CFR 803.17(a) (2) – Falta de un sistema para determinar los eventos reportables a la FDA (8 observaciones) El procedimiento escrito de MDR no incluye un sistema interno que permita un proceso estandarizado de revisión para determinar cuándo un evento cumple los criterios para ser reportado a la FDA.

- 21 CFR 803.17(a) (3)- Falta de un sistema para enviar los reportes a la FDA en el tiempo debido (8 observaciones) El procedimiento escrito de MDR no incluye un sistema interno que permita la transmisión oportuna de los reportes de MDR a la FDA.
- 21 CFR 803.17 (b) (1) – Falta de requerimientos de la información evaluada para determinar si un evento es reportable a la FDA (6 observaciones) El procedimiento escrito de MDR no incluye los requerimientos de documentación y mantenimiento de registros, para toda la información que fue evaluada para determinar si un evento es reportable.
- 21 CFR 803.17 (b) (2) – Deficiencias en los reportes y la documentación de la información (2 observaciones) El procedimiento escrito de MDR no incluye los requerimientos de documentación y mantenimiento de registros para todos los MDRs y para la información enviada a la FDA.
- 21 CFR 803.17 (b) (4)- Falta de información para facilitar el seguimiento oportuno por parte de la FDA (1 observación) El procedimiento escrito de MDR no incluye los requerimientos de documentación y mantenimiento de registros que permita el acceso a la información, y el seguimiento oportuno e inspección por parte de la FDA.
*******************************
En el siguiente blog de esta serie presentaremos el último subsistema del “Top Five” que recibió más observaciones de la FDA en el 2016: Controles de Producción y Proceso.

