





El uso de procedimientos escritos ayuda a estandarizar la ejecución de las actividades de manufactura, y evita la variabilidad a la hora de ejecutarlas. También aumenta la eficiencia de los empleados, proporcionando instrucciones claras y escritas de lo que deben hacer y cómo deben hacerlo. Finalmente, son una herramienta clave para los procesos de entrenamiento y facilita las labores de auditoría del Sistema de Calidad.
En esta entrada explicamos los requerimientos del apartado § 820.40 “Controles de Documentos” del CFR Parte 820, que aplican para la industria de Dispositivos Médicos, por parte de la FDA (Food & Drug Administration).
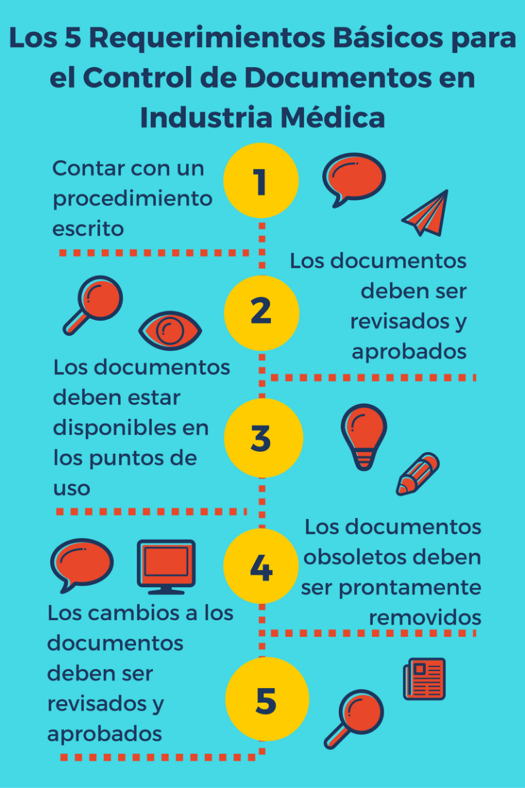
1. Tener un procedimiento escrito. La regulación requiere que tengamos un procedimiento donde se explique los mecanismos para controlar los documentos del Sistema de Calidad. Este procedimiento define todo el sistema de administración de los documentos, por ejemplo, si usamos un sistema electrónico, manual o mixto. En este procedimiento se define los requerimientos para los documentos, los cuales habitualmente son los siguientes:
Numero de revisión. Puede ser alfabético (A, B, C…) o numérico (01, 02, 03…). El número de revisión permite llevar el control de los cambios realizados al documento.
Fecha de emisión o efectividad. Es la fecha en que el documento se publica, ya sea de forma electrónica, o se postea de forma física, para ser usado.
Responsables de la aprobación. Indica quiénes tienen el nivel de autorización para aprobar el documento.
Alcance. Este apartado indica a qué áreas aplica el procedimiento y define qué funciones intervienen en la ejecución del mismo en cualquiera de sus fases.
Documentos y registros relacionados. Esta sección nos indica si al ejecutar un procedimiento debemos consultar otro documento relacionado, o completar un registro, para que quede evidencia de la realización de la actividad.

2. Los documentos deben ser revisados y aprobados. Antes de ser emitidos, los documentos deben ser revisados, para que sean exactos y adecuados para su uso, y aprobados. La aprobación debe incluir la fecha y firma de la persona que aprueba el documento, la cual debe ser un personal con un nivel técnico y autorización apropiada de acuerdo con la naturaleza del documento (por ejemplo un dueño de proceso o un experto técnico). Si usamos un sistema electrónico de control de documentos, es importante recordar que debemos cumplir con los requerimientos de la Parte 11, que regula las firmas electrónicas.
3. Disponibilidad de los documentos. Los documentos deben estar disponibles en todos los lugares o puntos de uso en los que se va a utilizar. Si tenemos un sistema electrónico de control de documentos, este puede considerarse como el punto de uso (si la mayoría de nuestro personal tiene acceso). Sin embargo, para áreas que no tengan un acceso directo al sistema, siempre es conveniente que se coloque de forma accesible una copia del documento. Por ejemplo, puede ser más “user friendly” para consulta rápida del personal en líneas de manufactura (workstations), áreas de bodega, recibo de materiales (receiving) o despacho de producto (shipping) el contar con los procedimientos de forma impresa.


5. Control de cambios. Los cambios a los documentos deben ser revisados y aprobados por personal con la misma función que realizó la aprobación inicial. Por ejemplo, si hacemos cambios a un procedimiento de “Inspección de Calidad de la Estación de Soldadura”, que fue aprobado en su emisión inicial por el Ingeniero de Calidad de la Estación de Soldadura, los cambios y revisiones subsecuentes deben ser aprobados por un Ingeniero de Calidad de esa misma área. Se debe mantener registros de los cambios a los documentos, incluyendo la descripción del cambio, los documentos afectados, la firma, fecha de aprobación, y cuándo el cambio se vuelve efectivo. Finalmente, los cambios a los documentos deben comunicarse al personal aplicable de forma oportuna.

