



Con acceso a internet, y a mercados globalizados y cada vez más competitivos, los clientes están más empoderados que nunca. Es por eso que en la actualidad, todo tipo de negocio (sea de manufactura o servicios), presta primordial importancia a las quejas de los clientes. Las mismas se responden y solucionan de la forma más inmediata y mejor posible; además se analizan e investigan para implementar mejoras en los procesos que permitan reducirlas y mantener a los clientes satisfechos.

En la industria de dispositivos médicos, las quejas de los clientes se investigan y responden no sólo por un sentido básico de negocio, sino además por requerimientos regulatorios, pues la FDA toma muy en serio las quejas de los dispositivos médicos que están en el mercado.
La parte 820.198 Registros de Quejas establece los requerimientos específicos de la agencia para que los manufacturadores reciban, evalúen e investiguen las quejas. Si usted es un profesional de calidad que trabaja o se relaciona con en el análisis de quejas, este blog le puede ser útil pues analizamos las principales observaciones de la FDA en el 2016.

En un blog anterior, presentamos “Las 5 principales observaciones de la FDA para dispositivos médicos durante el año fiscal 2016”. Con 409 observaciones, “Quejas” fue el tercer apartado de la regulación que recibió más 483s en el 2016. Adicionalmente, ha sido uno de los apartados de la regulación que más ha recibido observaciones en las inspecciones de la FDA en los últimos cinco años.
Interesantemente, el siguiente Pareto muestra que la gran mayoría de las observaciones fueron por deficiencias en los procedimientos de manejo de quejas.
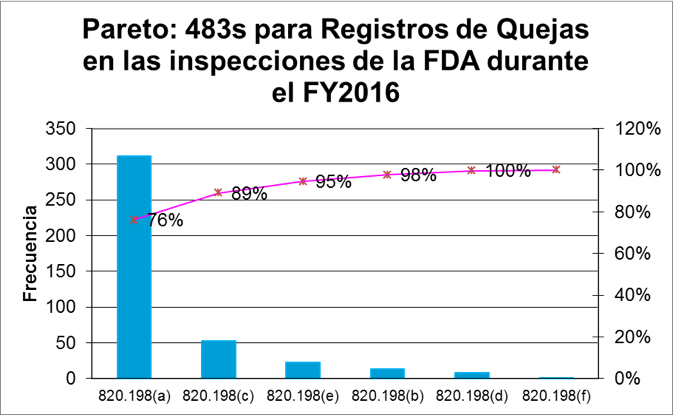 198(a) Procedimientos inadecuados– 311 observaciones (76%)
198(a) Procedimientos inadecuados– 311 observaciones (76%)
La gran mayoría de las observaciones para el proceso de quejas fueron por procedimientos inadecuados. La regulación indica que el manufacturador debe implementar procedimientos para recibir, revisar y evaluar las quejas recibidas (ya sea de forma electrónica, escrita u oral) por medio de una unidad formalmente designada (la cual puede ser un departamento o bien una sola persona).
Las observaciones recibidas fueron por procedimientos que no cumplían con todos los requerimientos de la regulación.
- 198(c) Quejas que requieren investigación no se investigaron – 53 observaciones (13%)
El apartado c) establece que todas las quejas que involucren una posible falla potencial del dispositivo, su empaque o su etiquetado, deben ser investigadas.
- 198(e) Registros de investigación deficientes – 23 observaciones (6%)

Este apartado establece que los registros de investigación de las quejas deben incluir:
- El nombre del dispositivo
- La identificación (si tuviera) y el número de control (mejor conocido como número de lote) del dispositivo
- La fecha de la queja
- El nombre, dirección y número de teléfono de la persona que puso la queja
- Los detalles de la queja
- La fecha y resultados de la investigación
- La acciones correctivas
- La respuesta de la queja
Las observaciones se dieron por deficiencias en la información requerida, o por registros faltantes.
- 198(b) Falta de evaluación adecuada de la queja para determinar si requiere investigación- 13 observaciones (3%)
No todas las quejas que recibe el manufacturador son válidas. El apartado b) indica que el manufacturador debe realizar una evaluación inicial de la queja para determinar si es necesaria una investigación. En el caso de no requerir investigación, debe documentase la razón (rationale) por la cual no es requerida, junto con el nombre de la persona responsable de tomar la decisión.
Las 13 observaciones en este apartado fueron porque no todas las quejas fueron evaluadas adecuadamente, o bien no se documentó adecuadamente la razón por la cual se decidió no investigar las quejas.
- 198(d) Falta de cumplimiento con los requerimientos de MDR – 8 observaciones (2%)
Hay un grupo especial de quejas que deben ser reportadas directamente a la FDA: las que están potencialmente relacionadas con la muerte de un paciente o una lesión seria. Estas quejas están reguladas por la parte 803 Medical Device Reporting (MDR).
Estas quejas deben ser prontamente evaluadas e investigadas por una persona designada; y mantenidas claramente identificadas en un registro aparte de los registros de quejas regulares. De forma adicional a la información requerida en el apartado d) (para las quejas regulares), el registro de los MDRs debe contener la siguiente información:
- Si el dispositivo falló con el cumplimiento de especificaciones
- Si el dispositivo estaba siendo usado para tratamiento o diagnóstico
- La relación del dispositivo con el evento adverso reportado
Las observaciones recibidas en este apartado fueron por incumplimiento de estos requerimientos, o bien por deficiencias en la información requerida en los registros de MDR.
820.198(f) Falta de accesibilidad de los registros- Sólo 1 observación
En ocasiones el departamento que recibe y revisa las quejas está en una localización separada de la planta de manufactura; por lo cual la regulación requiere que las plantas tenga acceso a los registros de quejas.
*******************************
En el siguiente blog presentaremos las observaciones de la FDA en el FY2016 para los Reportes de MDRs, el cual es un sistema complementario a la parte 820.198 Registros de Quejas, pues se ocupa de las quejas relacionadas con eventos adversos que deben reportarse a la FDA.